【关键词】 药品检查;药品变更;检查缺陷;变更管理;警告信
药品变更管理贯穿于药品全生命周期,以药品上市为节点,药品变更分为上市前变更与上市后变更。药品上市后变更管理是药品质量风险管理的重要内容,是国内外各类监督检查的重点之一。监管机构鼓励药品上市许可持有人(简称持有人)运用新技术、新方法、新设备、新科技成果,不断改进和优化生产工艺,持续提高药品质量,提升药品安全性、有效性和质量可控性,但不得对药品质量产生不良影响或带来潜在风险 [1]。近年来,在国内外药品检查中发现持有人在药品上市后变更管理方面仍有不足,例如变更控制管理体系建立不完善、变更管理类别不恰当、变更研究不充分等 [2-3]。因此,本文就我国药品上市后变更管理框架及常见问题进行系统分析。
一、药品变更概述
1.1 药品变更法规框架
自2019年新修订的《中华人民共和国药品管理法》颁布实施后,《药品注册管理办法》《药品生产监督管理办法》及《药品上市后变更管理办法(试行)》相继制修订并颁布实施,《药品注册申请审评期间变更工作程序(试行)》《已上市化学药品药学变更研究技术指导原则(试行)》《已上市化学药品和生物制品临床变更技术指导原则》等配套文件也陆续发布施行。浙江省、广东省、江西省等省级药品监督管理局(简称省药监局)陆续出台了本省和与上述变更文件相配套的文件,其中个别省药监局结合监管需要,进一步细化了变更情形和监管要求,例如江西省药监局于2023年6月出台《药品生产许可关键生产设施变化管理暂行规定》,将关键生产设施等条件变化实施登记管理制度,将变化情形细分为及时检查情形和纳入年度计划检查情形。至此,关于药品变更的工作程序和技术要求已基本明确,我国的药品变更法规体系基本完善,覆盖药品全生命周期的药品变更管理法规框架基本建成。部分药品变更法规文件见表1。
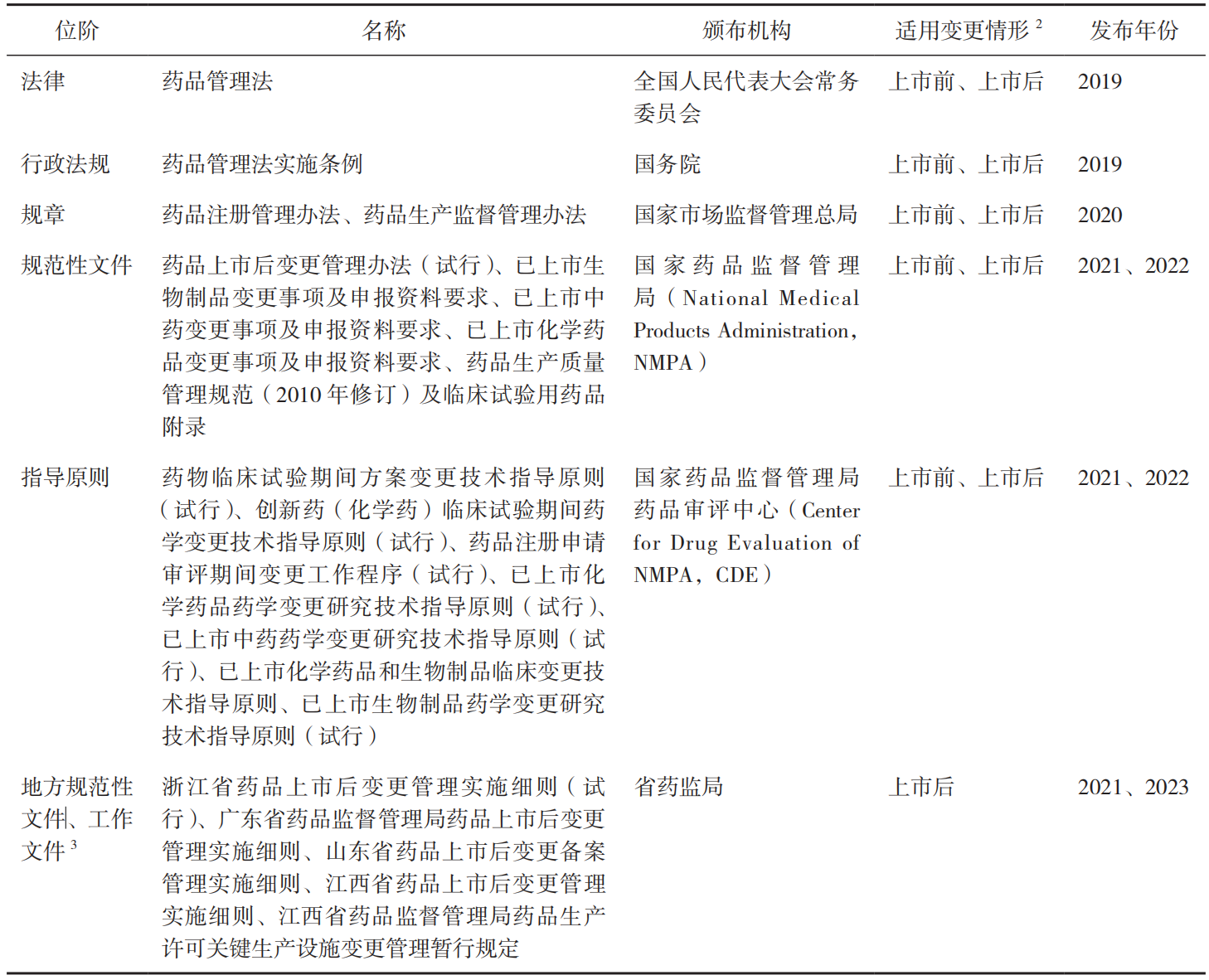
注:1 仅举例部分文件;2仅是列举主要适用的变更情形,上市前、上市后变更均可参考相应法规;3《浙江省药品上市后变更管理实施细则(试行)》发布年份为 2021 年,其余文件发布年份均为 2023 年。
1.2 药品变更管理框架
以药品上市为节点,药品变更分为上市前变更与上市后变更。药品上市前变更根据注册申报阶段的不同,分为药物临床试验申请和临床试验期间的补充申请审评期间变更、药品上市许可申请审评期间的变更,变更报CDE审评审批 [5]。
《药品上市后变更管理办法(试行)》将药品上市后变更分为注册管理事项变更和生产监管事项变更。注册变更管理类别根据法律法规要求和变更对药品安全性、有效性和质量可控性可能产生影响的风险程度,分为审批类变更、备案类变更和报告类变更,生产监管事项变更分为许可事项变更和登记事项变更 [6-7]。近年来,NMPA不断优化药品上市后变更监管工作程序,2024年2月NMPA印发优化药品补充申请审评审批程序改革试点工作方案,工作方案指出,以化学药品为重点,试点省级药品监管部门按照“提前介入、一企一策、全程指导、研审联动”的原则,为辖区内药品重大变更申报前提供前置指导、核查、检验和立卷服务 [8]。
对于不需主动告知监管部门,仅需在持有人内部药品质量体系中管理和记录的变更,将其称之为质量体系类变更,例如文件体系的变更、清洁和消毒方法的变更、组织架构的变更、公用系统局部改造等,质量体系类变更是贯穿药品上市前(技术转移、工艺验证、临床用样品制备等阶段)、上市后阶段,对于这类变更会在监管机构组织的常规检查或其他检查中作为重点检查项目之一 [9-10]。药品变更管理框架见图1。

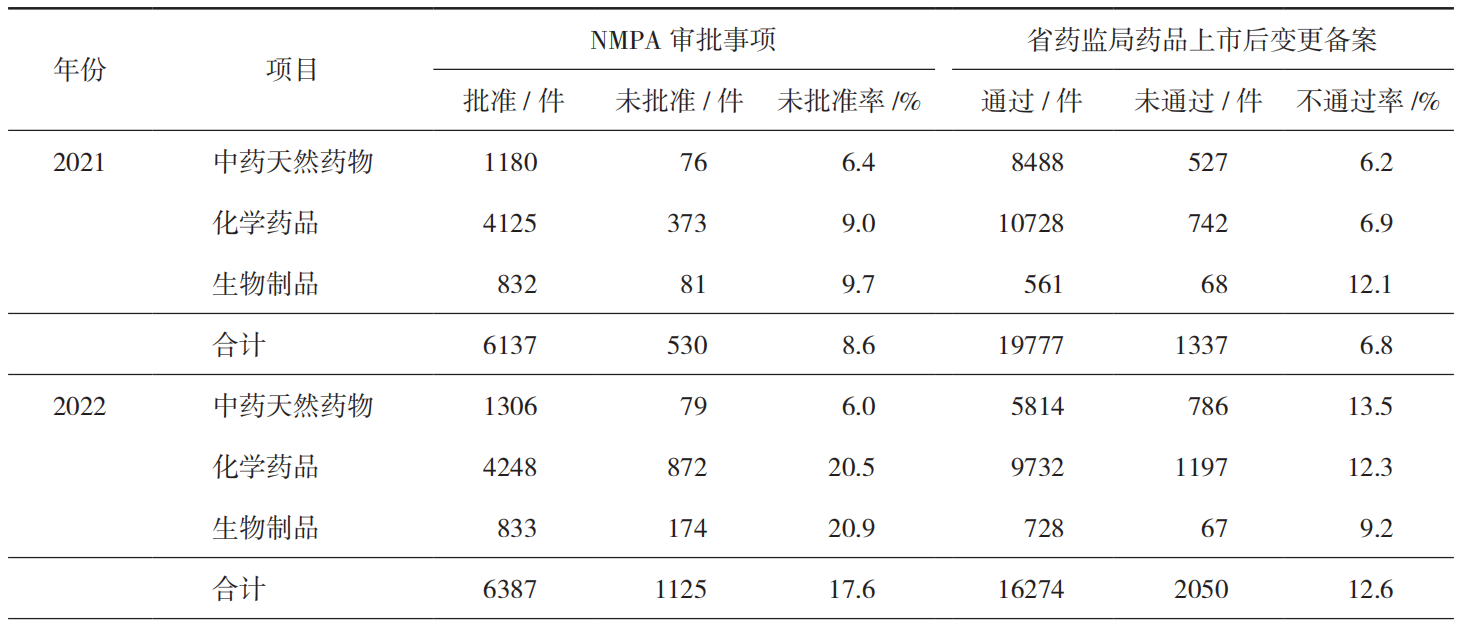
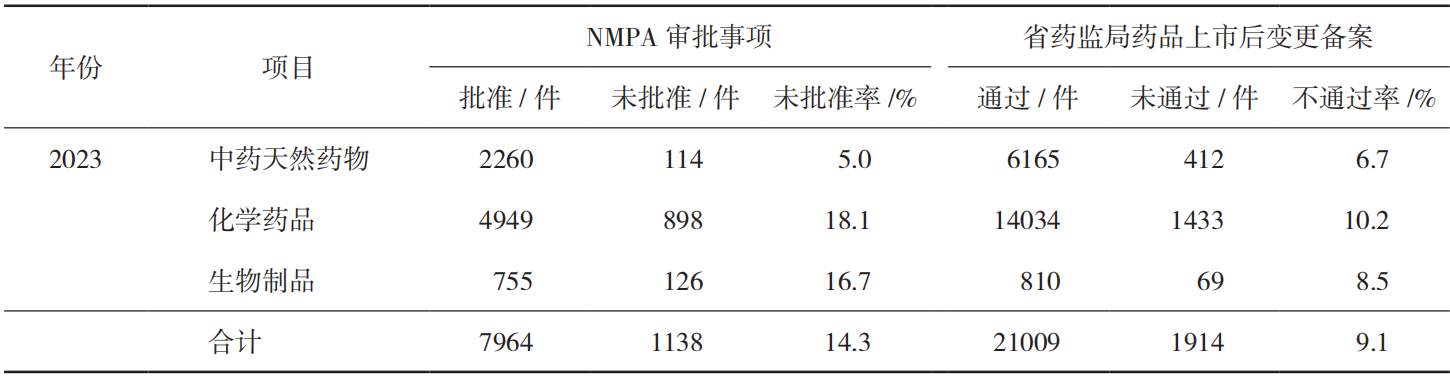
变更控制检查缺陷在各个国家、地区药品监督检查中经常出现。在2015年至2018年世界卫生组织(World Health Organization,WHO)对我国药品生产企业检查提出的各类缺陷中,“质量控制与质量保证”部分提出的缺陷数量最多,变更控制缺陷出现的频次较高,其常见问题主要包括变更管理程序规定不科学、未基于风险进行变更控制、变更风险评估不足及措施执行不全面、未执行变更程序等 [2]。
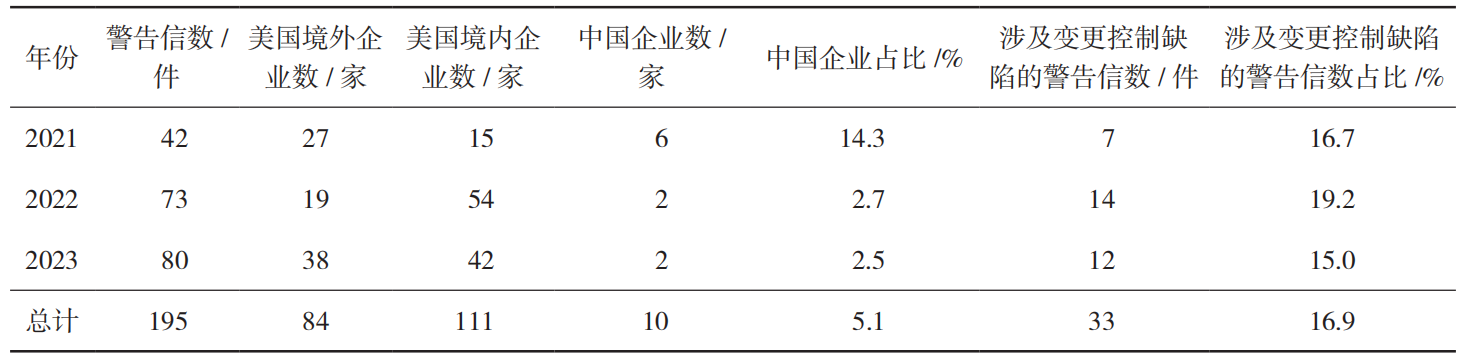
笔者以“Current Good Manufacture Practices”为关键词检索美国食品药品管理局(Food and Drug Administration,FDA)官网的警告信(Warning Letter)数据库,梳理2021年1月至2023年12月涉及现行药品生产质量管理规范(Current Good Manufacture Practices,CGMP)检查的警告信,共计195件 [16],其中出现变更控制缺陷的警告信共40件,占比20.5%,详见表3。CGMP检查的警告信主要由FDA下设的药品审评与研究中心(Center for Drug Evaluation and Research,CDER)及监管事务办公室(Office of Regulatory Affairs,ORA)内设的药品质量运营办公室(Office of Pharmaceutical Quality Operations,OPQO)发布,其中CDER主要发布美国境外药品生产企业CGMP警告信,OPQO发布美国境内药品生产企业CGMP警告信 [17]。
2.2.3 质量管理类变更常见问题
质量管理类变更不需主动告知监管部门,依靠内部变更控制程序,变更内容涉及药品生产质量方面,对应《药品生产质量管理规范(2010年修订)》的章节,主要涵盖机构与人员、厂房与设施、设备、物料与产品、文件管理、生产管理、质量控制与质量保证、委托生产与委托检验等方面内容 [26]。质量管理类变更常见问题类型主要包括以下3个方面。
(1)未纳入变更控制体系,未记录变更。这类问题常见的是未将设备变更,新增原辅料、包装材料供应商及变更包装标签样式(不改变内容)等情况纳入变更管理。具体案例:①口服固体制剂车间增加干法制粒机、方锥混合机设备及增加纯化水用水点,均未登记在当年变更台账中;②新增某原料药的经销商,但无相应的变更记录;③某颗粒外包装小盒的 [产品批号]、[生产日期]、[有效期至] 打印处底色由无底色改成黑色,未对该变更进行记录。
(2)未开展评估、确认、验证及相关研究工作。常见问题主要包括设备变更未进行设备确认,检验方法变更未进行验证或确认,变更生产工艺参数未进行相关验证等。具体案例:①固体制剂车间胶囊填充间新增加了一台全自动胶囊剂检重机,按照《变更控制管理规程》的要求提出了变更申请,但未对新增设备对产品质量的潜在影响进行评估和分析,未进行设备确认;②企业的医用氧标准根据NMPA修订件(批件号:XGB2021-061)进行了变更,制定了医用氧成品质量标准,但无方法确认记录;③企业根据《中华人民共和国药典》2020年版制法变更了酒黄精的工艺规程,但未进行工艺验证即开展商业化生产。
(3)文件变更管理不规范。除以上两种常见问题类型外,还有一类问题需关注,即文件变更,常见问题主要包括变更控制管理程序未根据NMPA发布的变更法规进行更新,以及变更管理程序设定不合理,文件变更未纳入变更控制体系,变更实施前相关文件未修订生效等。具体案例:①持有人《变更控制管理规程》未依据2021年NMPA发布的《药品上市后变更管理办法(试行)》及有关技术指导原则对相应的变更管理规程进行修订;②持有人《变更控制管理制度》列举的变更情形仅包括药学变更,未包括生产监管事项变更、临床变更、关键设施变化等情形;③某乳剂申请增加生产批量(微小变更),经3批工艺验证后,质量负责人审批同意新增生产批量,但相应工艺规程未修订。
三、结论与建议
我国药品变更管理框架基本完善,持有人在药品上市后变更管理方面仍有不足。持有人承担药品全生命周期的质量安全主体责任,是药品上市后变更管理的责任主体。对于持有人的药品上市后变更管理,笔者有如下几点建议。(1)持有人应在充分学习、理解药品变更法规基础上,建立科学合理的内部变更控制体系,制定完善的药品变更控制管理程序文件,制定符合法规要求并满足内部管理需求的内部变更分类原则,变更事项内容不仅应涵盖生产监管事项、注册管理事项,还应包括质量管理类变更。(2)应开展充分的变更研究工作,充分学习中药制剂、化学药品、生物制品药学和临床变更指导原则以及《已上市化学药品药学变更研究技术指导原则(试行)》溶出曲线研究、原料药变更的问答等技术文件,掌握变更研究的技术要求。(3)充分运用监管部门的沟通交流机制,对于尚不确定的变更分类,建议提前与药品监管部门沟通交流,并定期收集CDE、各省级药品监管部门公布的关于变更管理的共性问题解答。
药品检查是监管部门保证药品质量的重要监管措施,是发现持有人药品上市后变更问题的重要手段。药品检查员应注重知识管理和检查经验总结,形成系统全面的变更理念,对检查过程中涉及的药品上市后变更问题进行针对性分析,避免“一刀切”现象的出现。总之,持有人与监管部门均应懂得运用风险管理理念,深刻认识变更对产品质量的潜在影响,加强变更管理,确保药品质量。