本文选自:中国临床药理学杂志 第39卷 第7期1060-1064
作者简介:刘美霞,主管药师,主要从事药物技术审评工作
通信作者:王骏,研究员
作者单位:国家药品监督管理局 药品审评中心
化学药品改良型新药是在已知活性成分的基础上,对其结构、剂型、处方工艺、给药途径、适应证等进行优化,具有明显临床优势的药品。不同于全新靶点和结构创新药研发的高投入、长周期、高风险,改良型新药可借鉴已知活性成分药品的研究数据,降低研发成本、缩短临床研发的周期、提高临床成功率。随着制药工业技术的快速发展,改良型新药已成为当前新药研发的热点方向之一。改良型新药的评价通常基于整体证据,包括药学、非临床及临床研究内容。其中,临床药理研究对改良型新药的开发至关重要。目前,国内已发布《化学药品改良型新药临床试验技术指导原则》和《改良型新药调释制剂临床药代动力学研究技术指导原则》,尚未发布针对改良型新药临床药理研究的指导原则。国内已申报的化学药品改良型新药中,临床药理研究主要存在以下问题:①对于简单的不改变药代动力学(PK)特征的改剂型类改良型新药,试验过程中未验证临床优势,如口崩片的临床优势为在口腔中直接崩解后服用,无需用水送服,但实际试验过程中此类制剂往往未进行无水服药的相对生物利用度研究。②对于改变PK特征的改良型新药,在临床试验过程中,经常存在改良型新药与对照制剂的PK特征比对不充分,剂量探索研究缺失,食物效应、药物相互作用研究等外因对药物体内暴露影响的研究欠缺等问题,从而导致无法为改良型新药的临床用法用量提供充分的科学依据。本文主要结合美国食品药品监督管理局(FDA)已上市化学药品改良型新药研究实例及审评过程中遇到的问题,对化学药品改良型新药的临床药理研究提出几点思考,以期为国内化学药品改良型新药的研发提供一些参考。
1 昂丹司琼口溶膜
昂丹司琼原研企业为葛兰素史克,片剂(商品名:Zofran)于1992年获FDA批准上市,口崩片(商品名:Zofran ODT)于1999年获FDA批准上市。昂丹司琼临床上主要作为止吐药,用于由细胞毒性药物化疗和放射治疗引起的恶心呕吐,也可用于预防和治疗手术后的恶心呕吐。为了提高吞咽困难患者的顺应性,Par Pharmaceuticals, Inc.开发了昂丹司琼口溶膜(商品名:Zuplenz),并于2010年通过505b(2)途径在FDA获批上市。昂丹司琼口溶膜上市时开展的临床研究,见表1。
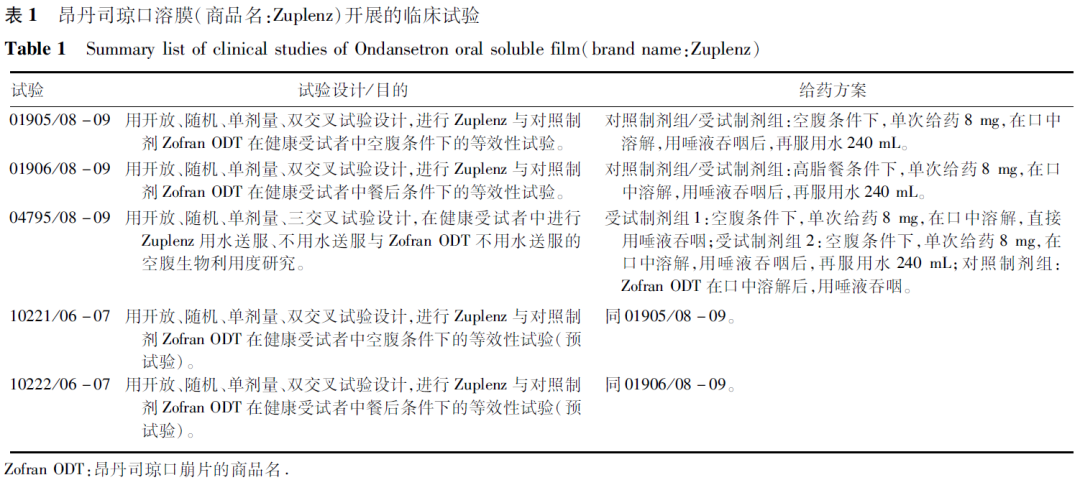
昂丹司琼口溶膜上市时主要通过空腹、餐后2项关键性生物等效性试验桥接原研昂丹司琼口崩片的安全有效性数据(含成人及儿科患者人群):昂丹司琼口溶膜与昂丹司琼口崩片在空腹、高脂餐后条件下,单次口服8 mg具有生物等效性,昂丹司琼口溶膜在舌头上溶解时间稍长于昂丹司琼口崩片,约快3~4
s。同时考察了昂丹司琼口溶膜不同服药方式对相对生物利用度的影响:在健康受试者空腹条件下,用水送服、无需水送服昂丹司琼片8 mg与无需水送服昂丹司琼口崩片8 mg具有生物等效性。
2 醋酸阿比特龙片
原研Janssen
Biotech的醋酸阿比特龙片(商品名:ZYTIGA)于2011年4月在美国获批上市,2015年获批进口国内。醋酸阿比特龙片在临床上主要与泼尼松或泼尼松龙合用,用于前列腺癌的治疗。醋酸阿比特龙片的推荐剂量为每日1 000 mg,单次服用;至少在服药前2 h内和服药后1 h内不能进食。醋酸阿比特龙片存在生物利用度低、体内暴露个体变异大且食物影响显著等问题。
Sun
Pharma Global, FZE的醋酸阿比特龙改良型新药于2018年5月通过505b(2)途径在FDA获批上市,商品名为YONSA,用SoluMatrix微粒技术制造工艺,提供了一种微粒化的醋酸阿比特龙片剂,使其在体内能被更有效地吸收,从而减少了临床使用剂量,降低了食物因素的影响。醋酸阿比特龙片(商品名:YONSA)推荐剂量为500 mg,每日1次,空腹或餐后口服。申报上市时开展的临床试验见表2,包括4项PK研究和1项基于睾酮抑制的PK/药效学(PD)研究。
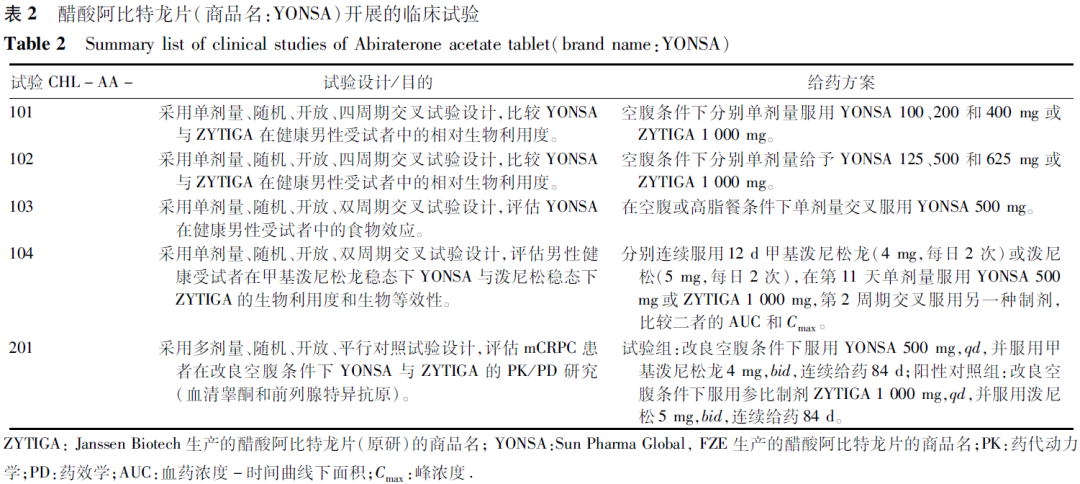
临床药理研究结果显示:阿比特龙在125~625
mg内,AUC呈剂量比例增加,Cmax呈略微低剂量比例增加。醋酸阿比特龙片(商品名:YONSA)与醋酸阿比特龙片(商品名:ZYTIGA)两制剂PK方面的差异主要在吸收和生物利用度方面。健康男性受试者单次给药醋酸阿比特龙片(商品名:YONSA) 500 mg与单次给药醋酸阿比特龙片(商品名:ZYTIGA)1 000 mg具有生物等效性。健康受试者空腹或高脂餐后单剂量服用醋酸阿比特龙片(商品名:YONSA) 500 mg,高脂餐后阿比特龙的Cmax和AUCinf是隔夜空腹条件下的6.5倍和4.4倍。稳态甲泼尼松龙/泼尼松下,空腹条件下单次给药醋酸阿比特龙片(商品名:YONSA) 500 mg与空腹单次给药醋酸阿比特龙片(商品名:ZYTIGA) 1 000 mg阿比特龙在人体内的系统暴露量相似。转移性去势抵抗性前列腺癌(mCRPC)患者在改良空腹条件下多次给药后的主要有效性终点――第9和10天平均血清睾酮水平,醋酸阿比特龙片(商品名:YONSA;n=24)和醋酸阿比特龙片(商品名:ZYTIGA;n=28)睾酮抑制情况相似;醋酸阿比特龙片(商品名:YONSA)在改良空腹条件下多次给药后的稳态暴露量低于醋酸阿比特龙片(商品名:ZYTIGA)。最终醋酸阿比特龙片(商品名:YONSA)基于与醋酸阿比特龙片(商品名:ZYTIGA)的PK、PD相似性在FDA获批上市。
综上,醋酸阿比特龙改良型新药目的为避免食物影响,增加生物利用度,降低服用剂量。在探索到目标制剂与对照制剂临床用药暴露量相当的剂量后,进行该剂量下本品相应用法下的最低和最高暴露量研究,并与对照制剂醋酸阿比特龙片(商品名:ZYTIGA)进行对比。醋酸阿比特龙片(商品名:YONSA)的暴露量上下限分别不超过后者暴露量的上下限或达到生物等效。
3 伊立替康脂质体注射液
伊立替康在人体组织内由非特异性羧酸酯酶转化为活性代谢物SN-38发挥主要治疗作用。SN-38通过关键代谢酶――尿苷二磷酸葡糖醛酸转移酶1A1(UGT1A1)转换为SN-38G(使用2种细胞系的体外细胞毒性测定中SN-38G的活性为SN-38的1/50至1/100)经胆汁及尿液排出。UGT1A1具有基因多态性,可导致SN-38葡糖醛酸化速率产生50倍差异。
普通盐酸伊立替康注射液及其冻干粉针制剂在体内易被代谢失活,采用脂质体剂型可以合理地改善伊立替康的PK和生物分布,同时保护其免于过早代谢。
Merrimack
Pharmaceuticals公司开发的伊立替康脂质体注射液(商品名:Onivyde;MM-398)于2015年通过505b(2)途径在FDA获批上市。伊立替康脂质体注射液在FDA申报上市时,开展的临床试验见表3,共包括6项与临床药理相关的研究及1项群体PK研究。

以上研究结果表明:伊立替康脂质体的直接测定表明,95%的伊立替康在循环过程中仍保持脂质体包封。与伊立替康注射剂(商品名:Camptosar)相比,输注MM-398后,总伊立替康的暴露量更高,SN-38的t1/2(3.0倍)和AUC0-inf(1.4倍)更高,但Cmax降低(0.19倍)。输注伊立替康脂质体注射液(商品名:Onivyde)、伊立替康注射剂(商品名:Camptosar)后,伊立替康转化为SN-38的转化率分别为2.89×10-4和1.50×10-2,SN-38转化为SN-38G的转化率分别为11.5和16.4,大部分的伊立替康以脂质体形式存在,限制了MM-398转化为SN-38。
群体PK研究结果表明:年龄、性别对伊立替康和SN-38的暴露没有临床意义的影响;在调整了体表面积(BSA)后,轻度到中度肾损害对总SN-38的暴露没有影响。基线总胆红素浓度为1~2 mg・dL-1(n=19)的患者总SN-38的平均稳态浓度比基线胆红素浓度<1 mg・dL-1(n=329)的患者增加了37%;对UGT1A1*28纯合子患者给予较低剂量伊立替康脂质体,该等位基因的纯合子(n=14)和非纯合子(n=244)患者的总SN-38平均稳态浓度分别为1.06和0.95 ng・mL-1;亚洲人总伊立替康平均稳态浓度比高加索人低56%,总SN-38平均稳态浓度高8%。
剂量-暴露-效应关系分析结果显示:在证明本品有效性的临床试验中,伊立替康脂质体+5-FU/LV治疗组患者的总伊立替康和SN-38血浆暴露水平升高与总生存期(OS)和无进展生存期(PFS)较长以及客观缓解率(ORR)较高相关。在353例患者的汇总分析中,血浆SN-38 Cmax较高与出现中性粒细胞减少症的可能性增加有关,血浆总伊立替康Cmax较高与出现腹泻可能性增加有关。
综上,伊立替康脂质体注射液通过采用脂质体剂型改善伊立替康的PK和生物分布,保护其免于过早代谢。伊立替康脂质体注射液的临床药理研究主要通过在患者中的单药剂量递增研究、联合用药的剂量递增研究获得本品的最大耐受剂量及初步PK特征;通过与普通制剂的比对研究进一步确认本品与普通制剂相比的PK行为差异;通过群体PK研究与暴露-效应关系分析为特殊人群(含基因多态性)给药剂量调整提供依据。
4 讨
论
结合FDA获批上市的昂丹司琼口溶膜、醋酸阿比特龙片、伊立替康脂质体注射液改良型新药的临床药理研究情况及同类品种国内申报情况,对化学药品改良型新药的临床药理研究提出以下几点思考。
对于普通的改剂型药物,如片剂、胶囊剂、分散片、颗粒剂、干混悬剂、口溶膜、无水吞服颗粒等普通口服剂型之间的剂型改变,通常不会改变药物的PK特征。对该类改良型新的临床药理研发思路,通常可通过与对照制剂进行相对生物利用度/生物等效性研究,桥接对照制剂的安全有效性数据。改良型新药与对照制剂的相对生物利用度/生物等效性研究同普通仿制药的生物等效性研究不同,研究过程中需验证目标制剂的临床优势,例如对于普通片改为口溶膜的改良型新药,口溶膜可在口腔内溶解后直接吞咽,提高吞咽困难患者的顺应性,在进行相对生物利用度/生物等效性研究的过程中,需考察口溶膜无水吞服给药方式下的相对生物利用度以及在口腔内的溶解时间等。通过优化制剂处方工艺等改变生物利用度的药物,如普通口服制剂改为调释口服制剂,普通静脉注射剂改为皮下给药注射剂等,该类改良型新药的临床药理研发思路,通常基于活性成分达到与对照制剂相似的暴露量,从而桥接对照制剂的安全有效性数据。在探索到目标制剂与对照制剂临床用药暴露量相当的剂量后,进一步考察外因如食物效应等因素对体内暴露行为的影响,为临床用法用量提供科学依据。
对于食物效应研究,通常建议头对头比较目标制剂在空腹、高脂餐条件下的体内暴露量与对照制剂获批的空腹和/或高脂餐条件下的体内暴露量差异,推荐采用三交叉或四交叉研究设计。对需要联合用药的药物,为考察联合用药对目标制剂与对照制剂的系统暴露量的影响,通常需开展联合用药后的相对生物利用度/生物等效性研究。
对于改变药物吸收及组织分布的改良型新药,如普通注射剂改为脂质体、微球等,通过改变活性药物的释放特征以及在机体的吸收、组织分布,导致药物在体内PK特征发生明显改变。对该类药物一般无法通过体内暴露量直接桥接对照制剂的安全、有效性数据,临床药理研究主要为探索目标药物的PK特征和剂量-暴露-效应关系,确立给药方案。进行该类药物与对照制剂的PK比对研究,阐述二者在吸收、分布、代谢和排泄方面的差异,有利于寻找最佳临床给药方案。此类药物可参照《化学药创新药临床单次和多次给药剂量递增PK研究技术指导原则》《创新药临床药理学研究技术指导原则》等进行相关研究。
需要说明的是,本文是基于目前的科学认知、背景调研等提出的化学药品改良型新药临床药理研究的相关考虑,不代表监管机构的要求。